Home > D. Systemic pathology > Genetic and developmental anomalies > conformational diseases
conformational diseases
Friday 15 April 2005
The conformation of proteins is maintained by the tight packing of their amino-acid side-chains; even small changes that result from the replacement of a single amino acid can be sufficient to allow conformational unfolding. This is especially true of some key domains of functional proteins, and is well illustrated by the serpins.
Many diseases arise from mutations that primarily affect neither expression nor function, but result in a protein with decreased conformational stability.
This conformational instability can intermittently cause the affected protein to unfold and then undergo intermolecular linkage, which results in intracellular aggregation that causes cumulative cell damage.
The insidious nature of protein accumulation explains why some familial diseases characteristically affect individuals in middle- or old-age.
The aggregation of conformationally destabilized proteins is now known to be a feature of many of the neurodegenerative diseases, notably of Alzheimer disease and Parkinson disease and the spongiform encephalopathies. (see serpins, serpin family of protease inhibitors).
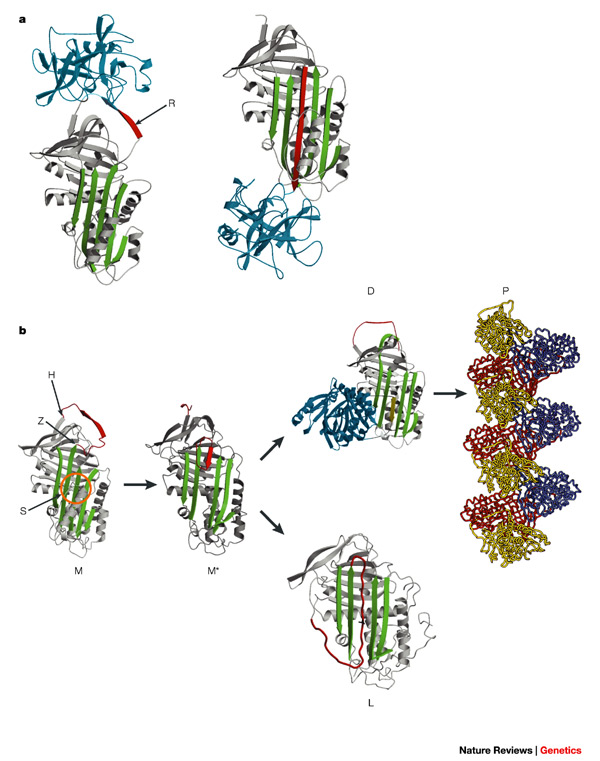
The common consequence of the mutations in all these diseases is the molecular instability of the encoded protein, which results in the formation of intermolecular beta-linkages, in which single peptide strands become aligned to form highly stable beta-sheets. These beta-linked structures then accumulate to cause cell death and hence disease.
Pathogenesis
Numerous diseases, including Alzheimer disease, Parkinson disease and other late-onset neurodegenerative diseases, arise from the conformationally driven aggregation of individual proteins.
Previous focus on just one end-product of such aggregation - extracellular deposits of amyloid - has diverted attention from what is now recognized as being primarily intracellular disease processes.
Recent structural findings show how cytotoxicity can result from even minor changes in conformation that do not lead to amyloid formation, as with the accumulation within the endoplasmic reticulum of intact mutant alpha-1-antitrypsin in hepatocytes and of neuroserpin in neurons.
Studies in Alzheimer disease and other dementias also indicate that the damage occurs at the stage of the initial intermolecular linkages that precede amyloid formation.
A disadvantageous gain of function
A defining characteristic of conformational diseases is that they arise from mutations that result in a disadvantageous gain of function. Such mutations have consequences over and above any accompanying loss of function.
So, the liver cirrhosis that is associated with alpha1-antitrypsin deficiency arises from conformationally destabilizing mutations of alpha1-antitrypsin, but not from the null mutations that result in total non-expression.
Similarly, whereas non-expressing null variants of antithrombin (AT3) result in a predisposition to thrombosis, an even greater threat of thrombosis occurs with conformationally destabilizing variants.
These carry with them, as well as plasma deficiency, the risk of pathological monomeric and polymeric conformational transitions that inactivate the protein, with an accompanying sudden and severe onset of thrombosis.
Therapeutic pathways
The realization that the conformational diseases are due to aberrant beta-linkages opens the possibility of general as well as specific approaches to treatment. Current attempts are aimed at either blocking the formation of beta-linkages or increasing the turnover of the accumulated aggregates.
These approaches are typified by continuing studies that aim to block the linkage between the reactive-centre loop and beta-sheet A, which underlies the polymerization of the serpins.
The polymerization of Z alpha1-antitrypsin can be blocked by annealing reactive-loop peptides to beta-sheet A. These peptides were 11-13 residues long and able to anneal to other members of the serpin superfamily. This was most clearly shown by the finding that a reactive-loop peptide of antithrombin annealed more readily to beta-sheet A of alpha1-antitrypsin and vice versa.
These peptides, although useful in establishing the mechanism of polymerization, are too long and too promiscuous to be used for rational drug design.
Chemical chaperones can also be used to stabilize intermediates of the folding pathway.
The chaperone trimethyamine oxide has no effect on the secretion of Z alpha1-antitrypsin in cell culture as it allowed the conversion of unfolded Z alpha1-antitrypsin to polymers.
By contrast, the chaperone 4-phenylbutyrate (4-PBA) increases the secretion of Z alpha1-antitrypsin from cell lines and transgenic mice.
This agent has been used for several years to treat children with urea cycle disorders and more recently 4-PBA has been shown to increase the expression of mutant (deltaF508) cystic fibrosis transmembrane regulator protein in vivo.
These encouraging findings have led to a pilot study being carried out to evaluate the potential of 4-PBA to promote the secretion of alpha1-antitrypsin in patients with alpha1-antitrypsin deficiency.
The blockage of the beta-strand linkage that underlies the serpinopathies need not be complete, but merely sufficient, to allow the cell to degrade the aberrant protein.
This is also likely to be the case for beta-strand blocking agents used to prevent the other beta-linked conformational diseases.
Each disease will require a different beta-strand blocking agent targeted to the appropriate organelle of the cell or the extracellular matrix. is being illustrated by study of the serpins.
Types
![]() alpha1-antitrypsin deficiency
alpha1-antitrypsin deficiency![]() conformational dementias
conformational dementias
- late-onset dementia
- amyloidoses
- prion encephalopathies
- Huntington disease
- Alzheimer disease
See also
![]() conformational diversity
conformational diversity
References
![]() Carrell RW. Cell toxicity and conformational disease. Trends Cell Biol. 2005 Nov;15(11):574-80. PMID: 16202603
Carrell RW. Cell toxicity and conformational disease. Trends Cell Biol. 2005 Nov;15(11):574-80. PMID: 16202603
![]() Bernier V, Lagace M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab. 2004 Jul;15(5):222-8. PMID: 15223052
Bernier V, Lagace M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab. 2004 Jul;15(5):222-8. PMID: 15223052
![]() Lomas DA, Carrell RW. Serpinopathies and the conformational dementias. Nat Rev Genet. 2002 Oct ;3(10):759-68. PMID : 12360234
Lomas DA, Carrell RW. Serpinopathies and the conformational dementias. Nat Rev Genet. 2002 Oct ;3(10):759-68. PMID : 12360234
![]() Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency: a model for conformational diseases. N Engl J Med. 2002 Jan 3;346(1):45-53. PMID: 11778003
Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency: a model for conformational diseases. N Engl J Med. 2002 Jan 3;346(1):45-53. PMID: 11778003
![]() Seidah NG, Prat A. Precursor convertases in the secretory pathway, cytosol and extracellular milieu. Essays Biochem. 2002;38:79-94. PMID: 12463163
Seidah NG, Prat A. Precursor convertases in the secretory pathway, cytosol and extracellular milieu. Essays Biochem. 2002;38:79-94. PMID: 12463163
![]() Chow MK, Lomas DA, Bottomley SP. Promiscuous beta-strand interactions and the conformational diseases. Curr Med Chem. 2004 Feb;11(4):491-9. PMID: 14965229
Chow MK, Lomas DA, Bottomley SP. Promiscuous beta-strand interactions and the conformational diseases. Curr Med Chem. 2004 Feb;11(4):491-9. PMID: 14965229
![]() Carrell RW, Lomas DA. Conformational disease.Lancet. 1997 Jul 12;350(9071):134-8. PMID: 9228977
Carrell RW, Lomas DA. Conformational disease.Lancet. 1997 Jul 12;350(9071):134-8. PMID: 9228977
{kind=link}